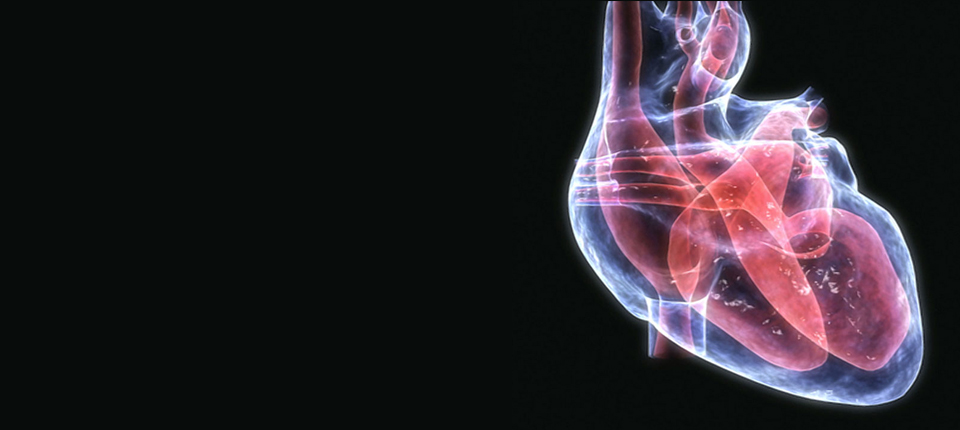
Blood in need of oxygenation flows into the right atrium, down to the right ventricle to be pumped through the pulmonary artery to the lungs.
When that blood comes back to the heart, chock full of oxygen, it arrives in the left atrium, down to the left ventricle to be pumped up through the aorta to the rest of the body before returning to the heart.
It's a cycle that happens billions of times in the average lifetime.
It's a coordination we admire and appreciate. Supporting that beat is essential to the work we do at Abbott. With February designated as Heart Health Month in the U.S., it takes on even greater significance to us.
"Research and development within the cardiovascular space has never been more exciting, and teams throughout Abbott are finding new ways to improve existing technology while also driving entire therapy segments in new directions," said Mark Strong, divisional vice president of program development within Abbott’s Cardiac Arrhythmias and Heart Failure division.
Here's just a sample of our expanding cardiovascular portfolio to care for your heart:
"As a leader in cardiovascular therapy development, we have to stay vigilant in capitalizing on new data, new technology and discoveries to optimize the devices and therapies across our entire portfolio to make sure that we're improving patients' lives," Strong said.
"In addition to pushing new technology forward, we have to make sure that each solution we bring to market is generating additional benefits, such as reducing the cost of care, improving workflow or incorporating new technology into how physicians and patients utilize medical devices. The problems we're trying to solve are massive, but the energy we're putting toward solving those problems is even greater."
Abbott takes seriously its job to provide high quality technologies that improve lives by providing knowledge to patients and tools to doctors. Our 103,000-strong company employs more than 2,300 scientists.
The sooner we can help get you start feeling better, the sooner you can get back to living your fullest, healthiest life.
"I'm just really encouraged by the possibilities," Jacobs said.
Taking care of your heart? It's in our blood.
References
*Abbott data on file.
AP2943687-WBU Rev. A
INDICATIONS, SAFETY & WARNINGS
RX ONLY
ENSITE PRECISION™ CARDIAC MAPPING SYSTEM
THE XIENCE SIERRA STENT SYSTEM
MITRACLIP CLIP DELIVERY SYSTEMS
AMPLATZER™ PFO OCCLUDER
SJM MASTERSSERIESMECHANICALHEARTVALVE MECHANICAL HEART VALVE SIZER
CONFIRM RX™
Model DM3500
Insertable Cardiac Monitor
INDICATIONS:
The Confirm Rx™ ICM is indicated for the monitoring and diagnostic evaluation of patients who experience unexplained symptoms such as: dizziness, palpitations, chest pain, syncope, and shortness of breath, as well as patients who are at risk for cardiac arrhythmias. It is also indicated for patients who have been previously diagnosed with atrial fibrillation or who are susceptible to developing atrial fibrillation. The Confirm Rx™ ICM has not been specifically tested for pediatric use.
CONTRAINDICATION:
There are no known contraindications for the insertion of the Confirm Rx™ ICM. However, the patient’s particular medical condition may dictate whether or not a subcutaneous, chronically inserted device can be tolerated.
ADVERSE EVENTS:
Possible adverse events (in alphabetical order) associated with the device, include the following: Allergic reaction, Bleeding, Chronic nerve damage, Erosion, Excessive fibrotic tissue growth, Extrusion, Formation of hematomas or cysts, Infection, Keloid formation and Migration. Refer to the User’s Manual for detailed indications, contraindications, warnings, precautions and potential adverse events.
ADDITIONAL INFORMATION:
Clinicians must log onto Merlin.net™ Patient Care Network to view transmissions from patients’ Confirm Rx™ ICM. On Merlin.net™ PCN they can configure transmission schedules and enable or disable features on a patient’s myMerlin™ mobile app. Review of transmissions is dependent on the clinician and may not happen immediately following delivery of such transmissions.
LIMITATION:
Patients may use their own Apple‡ or Android‡ mobile device to transmit information from their Confirm Rx™ ICM using the myMerlin™ mobile app. To do so the device must be powered on, app must be installed, Bluetooth® wireless technology enabled and data coverage (cellular or WiFi‡) available. The myMerlin™ mobile app provides periodic patient monitoring based on clinician configured settings. Data is resent if the transmission was not sent successfully. However, there are many internal and external factors that can hinder, delay, or prevent acquisition and delivery of ICM and patient information as intended by the clinician. These factors include: patient environment, data services, mobile device operating system and settings, ICM memory capacity, clinic environment, schedule/configuration changes, or data processing.
An Abbott mobile transmitter is available for patients without their own compatible mobile device.
RX ONLY
BRIEF SUMMARY:
Prior to using these devices, please review the Instructions for Use for a complete listing of indications, contraindications, warnings, precautions, potential adverse events and directions for use.
INDICATION FOR USE:
The CardioMEMS™ HF System is indicated for wirelessly measuring and monitoring pulmonary artery (PA) pressure and heart rate in New York Heart Association (NYHA) Class III heart failure patients who have been hospitalized for heart failure in the previous year. The hemodynamic data are used by physicians for heart failure management and with the goal of reducing heart failure hospitalizations.
CONTRAINDICATIONS:
The CardioMEMS™ HF System is contraindicated for patients with an inability to take dual antiplatelet or anticoagulants for one month post implant.
POTENTIAL ADVERSE EVENTS:
Potential adverse events associated with the implantation procedure include, but are not limited to the following: Infection, Arrhythmias, Bleeding, Hematoma, Thrombus, Myocardial infarction, Transient ischemic attack, Stroke, Death, and Device embolization.
Refer to the Instructions for Use for detailed indications, contraindications, warnings, precautions and potential adverse events.
ENSITE PRECISION™ CARDIAC MAPPING SYSTEM
CAUTION:
This product is intended for use by or under the direction of a physician. Prior to use, reference Instructions for Use, inside the product carton (when available) or at eifu.abbottvascular.com or at manuals.sjm.com for more detailed information on Indications, Contraindications, Warnings, Precautions and Adverse Events.
INDICATIONS:
The EnSite Precision™ Cardiac Mapping System is a suggested diagnostic tool in patients for whom electrophysiology studies have been indicated. The EnSite Precision™ System interfaces to either the MediGuide™ Technology System or the EnSite Precision™ Module to combine and display magnetic processed patient positioning and navigation mapping information. When used with the EnSite™ Array™ Catheter, the EnSite Precision™ Cardiac Mapping System is intended to be used in the right atrium of patients with complex arrhythmias that may be difficult to identify using conventional mapping systems alone. OR, when used with an EnSite Precision™ Surface Electrode Kit, the EnSite Precision™ Cardiac Mapping System is intended to display the position of conventional electrophysiology (EP) catheters in the heart.
WARNINGS:
Refer to the ablation catheter labeling for a listing of adverse events related to the use of this device in conjunction with radio frequency ablation, as a part of the diagnosis and treatment of cardiac arrhythmias. For patient safety, any connections that directly connect the patient to the EnSite Precision Cardiac Mapping System must be routed through the appropriate module: EnSite Precision Link, Sensor Enabled™ NavLink, EnSite Precision Field Frame, ArrayLink, CathLink, SJM ECG Cable, RecordConnect, or GenConnect. When using the EnSite Precision Module full protection against the effects of cardiac defibrillator discharge and other leakage currents is dependent upon the use of appropriate cables. Refer to the ablation catheter labeling for a listing of adverse events related to the use of this device in conjunction with radio frequency ablation, as a part of the diagnosis and treatment of cardiac arrhythmias. For patient safety, any connections that directly connect the patient to the EnSite Precision Cardiac Mapping System must be routed through the appropriate module: EnSite Precision Link, Sensor Enabled™ NavLink, EnSite Precision Field Frame, ArrayLink, CathLink, SJM ECG Cable, RecordConnect, or GenConnect. When using the EnSite Precision Module full protection against the effects of cardiac defibrillator discharge and other leakage currents is dependent upon the use of appropriate cables.
PRECAUTIONS:
Do not operate the EnSite Precision Field Frame within 10 meters (m) of another operating Field Frame. Do not place the EnSite Precision Field Frame cable inside the measurement volume or wrap it around the Field Frame, as it may create a magnetic interference. Do not coil the EnSite Precision Field Frame cable. The cable carries enough electric current that a magnetic field will be created when the cable is placed in a circular formation. This magnetic field may disturb the Field Frame’s magnetic field. Do not place the EnSite Precision Link, Sensor Enabled™ within 1 m of the EnSite Precision Field Frame - Do not place tool cables within 30 millimeters (mm) of the EnSite Precision Field Frame cable. If placed this close—particularly if the cables are parallel to each other—the tool cable may become subject to electromagnetic interference. Metallic equipment used in close proximity to the magnetic field during the procedure, such as a sterile drape holder, may affect Sensor Enabled (SE) points and SE field scaling accuracy. Do not use the EnSite Precision Cardiac Mapping System in the presence of other magnetic fields. Do not drop the EnSite Precision Field Frame or subject it to impact. Physical damage to the Field Frame may alter the Field Frame’s factory calibration.
THE XIENCE SIERRA STENT SYSTEM
The XIENCE V®, XIENCE nano®, XIENCE PRIME®, XIENCE PRIME® LL, XIENCE Xpedition®, XIENCE Xpedition® SV and XIENCE Xpedition® LL , XIENCE Alpine®, and XIENCE SierraTM (XIENCE Family) of Everolimus Eluting Coronary Stents on the MULTI-LINK VISION® or MULTI-LINK MINI VISION® Delivery System
INDICATIONS
The XIENCE Sierra stent system is indicated for improving coronary artery luminal diameter in patients, including those with diabetes mellitus, with symptomatic heart disease due to de novo native coronary artery lesions (length ≤ 32 mm) with reference vessel diameters of ≥ 2.25 mm to ≤ 4.25 mm. In addition, the XIENCE Sierra stent system is indicated for treating de novo chronic total coronary occlusions.
CONTRAINDICATIONS
The XIENCE Sierra stent system is contraindicated for use in:
WARNINGS
PRECAUTIONS
POTENTIAL ADVERSE EVENTS
Adverse events (in alphabetical order) which may be associated with percutaneous coronary intervention treatment procedures and the use of a coronary stent in native coronary arteries include, but are not limited to, the following:
The risks described below include, but are not limited to, the anticipated adverse events relevant for the cardiac population referenced in the contraindications, warnings, and precautions sections of the everolimus labels.
MITRACLIP CLIP DELIVERY SYSTEMS
INDICATION FOR USE
CONTRAINDICATIONS
The MitraClip™ NTR/XTR System is contraindicated in patients with the following conditions:
WARNINGS
PRECAUTIONS
POTENTIAL COMPLICATIONS AND ADVERSE EVENTS
The following ANTICIPATED EVENTS have been identified as possible complications of the MitraClipTM procedure.
Death; Allergic reaction (anesthetic, contrast, Heparin, nickel alloy, latex); Aneurysm or pseudo-aneurysm; Arrhythmias; Atrial fibrillation; Atrial septal defect requiring intervention; Arterio-venous fistula; Bleeding;
Cardiac arrest; Cardiac perforation; Cardiac tamponade / Pericardial Effusion; Chordal entanglement / rupture; Coagulopathy; Conversion to standard valve surgery; Deep venous thrombus (DVT); Dislodgement of previously implanted devices; Dizziness; Drug reaction to anti-platelet / anticoagulation agents / contrast
media; Dyskinesia; Dyspnea; Edema; Emboli (air, thrombus, MitraClipTM Implant); Emergency cardiac surgery; Endocarditis; Esophageal irritation; Esophageal perforation or stricture; Failure to deliver MitraClipTM to the intended site; Failure to retrieve MitraClipTM System components; Fever or hyperthermia; Gastrointestinal bleeding or infarct; Hematoma; Hemolysis; Hemorrhage requiring transfusion; Hypotension / hypertension; Infection; Injury to mitral valve complicating or preventing later surgical repair; Lymphatic complications; Mesenteric ischemia; MitraClipTM Implant erosion, migration or malposition; MitraClipTM Implant thrombosis; MitraClipTM System component(s) embolization; Mitral stenosis; Mitral valve injury; Multi-system organ failure; Myocardial infarction; Nausea / vomiting; Pain; Peripheral ischemia; Prolonged angina; Prolonged ventilation; Pulmonary congestion; Pulmonary thrombo-embolism; Renal insufficiency or failure; Respiratory failure / atelectasis / pneumonia; Septicemia; Shock, Anaphylactic or Cardiogenic; Single leaflet device attachment (SLDA); Skin injury or tissue changes due to exposure to ionizing radiation; Stroke or transient ischemic attack (TIA); Urinary tract infection; Vascular trauma, dissection or occlusion; Vessel spasm; Vessel perforation or laceration; Worsening heart failure; Worsening mitral regurgitation; Wound dehiscence
AMPLATZER™ PFO OCCLUDER
INDICATIONS AND USAGE
The AMPLATZER™ PFO Occluder is indicated for percutaneous transcatheter closure of a patent foramen ovale (PFO) to reduce the risk of recurrent ischemic stroke in patients, predominantly between the ages of 18 and 60 years, who have had a cryptogenic stroke due to a presumed paradoxical embolism, as determined by a neurologist and cardiologist following an evaluation to exclude known causes of ischemic stroke.
CONTRAINDICATIONS
Patients with anatomy in which the AMPLATZER™ PFO device size required would interfere with other intracardiac or intravascular structures, such as valves or pulmonary veins.
WARNINGS
PRECAUTIONS
ADVERSE EVENTS
Potential adverse events that may occur during or after a procedure using this device may include, but are not limited to: Air embolus Allergic drug reaction; Allergic dye reaction; Allergic metal reaction: Nitinol (nickel, titanium), platinum/iridium, stainless steel (chromium, iron, manganese, molybdenum, nickel); Anesthesia reactions; Apnea; Arrhythmia; Bacterial endocarditis; Bleeding ; Brachial plexus injury; Cardiac perforation; Cardiac tamponade; Cardiac thrombus; Chest pain; Device embolization; Device erosion; Deep vein thrombosis; Death; Endocarditis; Esophagus injury; Fever; Headache/migraine; Hypertension/hypotension; Myocardial infarction; Pacemaker placement secondary to PFO device closure; Palpitations; Pericardial effusion; Pericardial tamponade; Pericarditis; Peripheral embolism; Pleural effusion; Pulmonary embolism; Reintervention for residual shunt/device removal; Sepsis; Stroke; Transient ischemic attack; Thrombus; Valvular regurgitation; Vascular access site injury; Vessel perforation
SJM MASTERSSERIESMECHANICALHEARTVALVE MECHANICAL HEART VALVE SIZER
INDICATIONS FOR USE
The SJMTM Masters Series Mechanical Heart Valve is intended for use as a replacement valve in patients with a diseased, damaged, or malfunctioning mitral or aortic heart valve. This device
may also be used to replace a previously implanted mitral or aortic prosthetic heart valve.
The sizer model 905-15 is indicated to confirm size selection of the 15AHPJ-505 and 15MHPJ-505 valves.
CONTRAINDICATIONS
The SJMTM Masters Series Mechanical Heart Valve is contraindicated for individuals unable to tolerate anticoagulation therapy.
The sizer model 905-15 is contraindicated for use with any devices other than the 15 AHPJ-505 and 15MHPJ-505 valves. Any sizer sterilization method other than steam is contraindicated.
WARNINGS
PRECAUTIONS

Please be aware that the website you have requested is intended for the residents of a particular country or region, as noted on that site. As a result, the site may contain information on pharmaceuticals, medical devices and other products or uses of those products that are not approved in other countries or regions.
The website you have requested also may not be optimized for your specific screen size.


FOLLOW ABBOTT